Summary
This study uses total RNA-sequencing of Lyme disease patients’ peripheral blood mononuclear cells. Using novel data analysis approaches, three unique groups of Lyme disease patients were identified, each with characteristic clinical and immunological features. Data from healthy controls and COVID-19 patients were included. Using machine learning classifiers, the study shows that Lyme patients can be distinguished from healthy controls as well as from COVID-19 patients.
Why was this study done?
In the absence of a reliable laboratory diagnostic tool, the diagnosis of early Lyme disease depends on a demonstrated erythema migrans rash that occasionally does not present or is not observed. This can lead many patients to progress to the early disseminated stage and its debilitating symptoms before the disease is diagnosed and treated.
To further the understanding of the molecular mechanisms that lead to Lyme disease symptoms, the longitudinal changes in immune responses and gene expression were examined as a potential tool for characterizing Lyme disease patients, COVID-19 patients, and healthy controls.
How was this study done?
In the absence of a reliable laboratory diagnostic tool, the diagnosis of early Lyme disease depends on a demonstrated erythema migrans rash that occasionally does not present or is not observed. This can lead many patients to progress to the early disseminated stage and its debilitating symptoms before the disease is diagnosed and treated.
To further the understanding of the molecular mechanisms that lead to Lyme disease symptoms, the longitudinal changes in immune responses and gene expression were examined as a potential tool for characterizing Lyme disease patients, COVID-19 patients, and healthy controls.
What were the major findings?
The study showed that Lyme disease patients can be distinguished from the controls as well as from COVID-19 patients, but classification was not successful in distinguishing those patients with early Lyme disease cases that would advance to develop post-treatment persistent symptoms.
All Lyme disease cases exhibited gene expression features suggesting long-lasting immune changes, even those that returned to health.
Using immune profiling, thirty-eight cytokines and chemokines were measured in the sera of cases and controls and 5 were statistically significant by cluster group (p < 0.05, GM-CSF, IL-9, IL-12, MIP1β, IL-31), while 7 were borderline statistically significant (0.10 < p < 0.05: IL-23, IL-7, IL-13, IL-15, PDGF-ββ, IL-21, IL-17F).
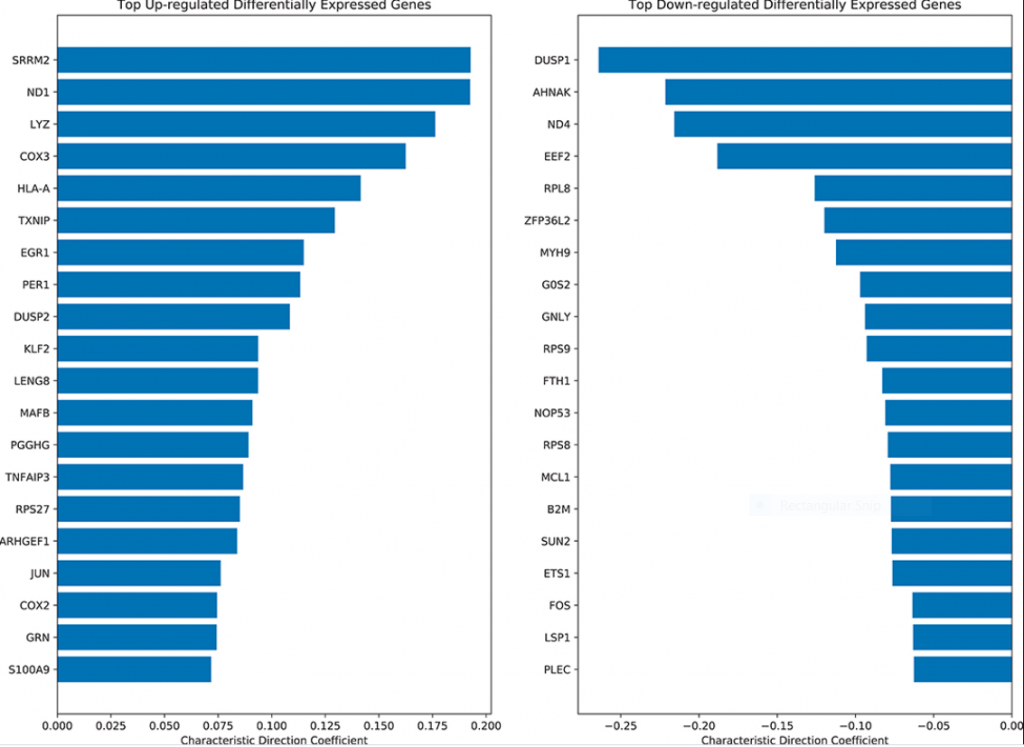
Through differential gene expression analysis specific immune response genes were shown to be upregulated in Lyme disease. The top-ranked up-regulated genes in Lyme disease cases were the human leukocyte antigens HLA-A and HLA-B, which suggest inflammation via adaptive immune activation as well as an association with arthritis pathways. Genes specific for diseases displaying erythema were also upregulated. The increase in immune response genes and dysregulation of gene expression continued for months post infection and treatment.
Analysis of different clusters showed distinctions in clinical characteristics and immunological features. It was observed that the Lyme disease cases had an activation of an immune response with significantly more features linked to T regulatory (Treg) cells, more monocytes, and less resting T memory cells.
What is the impact of this work?
The molecular mechanisms underlying the disease course and outcomes of Lyme disease are still poorly understood. This study reinforces the idea the human Lyme disease is a heterogeneous disease that likely involves diverse mechanisms. The performance of the study’s machine learning classifiers suggests that they could potentially aid in diagnosis. Additional information about the population of Lyme disease patients is needed to adequately assess the performance of this approach in a routine clinical setting. In addition, more samples and uniform processing of the data is required to determine if profiling host gene expression may be a useful and potentially sensitive approach for the diagnosis of acute Lyme disease.
The significance of the elevated Treg signal in this study of acute Lyme disease is not clear. Several studies have shown that in human tuberculosis increased Treg responses are associated with active disease and may impair pathogen clearance and disease resolution. Whether this is the case in some stages of human Lyme disease, or if these cells contribute to the dysregulated or maladaptive immune response in Lyme disease, will require additional studies with single cell RNA-sequencing.
It is unclear what is driving the persistent long-term alteration of genes in Lyme disease patients, but possibilities include the continued presence of Borrelia burgdorferi or foreign Bb antigens and/or epigenetic changes that could have been triggered by the infection. Identifying immune response patterns may help define Lyme disease subgroups and help inform treatment strategies. The cost of RNA-seq is continually dropping so it could become a practical approach in a clinical setting. Future studies are needed to better understand the differentiated immune mechanisms for multiple Lyme disease groups and comparisons with other infections, to determine the potential value of using detailed immune profiling in Lyme disease diagnostics and treatments.
This research was supported by:
RDRCC / P30
This research was funded by:
The project was partially supported by funds from the Cohen Lyme & Tickborne Disease Initiative, NIH grants U54HL127624, U24CA224260, and P30AR070254.
Study team members:
Daniel J. B. Clarke1†, Alison W. Rebman2†, Allison Bailey1, Megan L. Wojciechowicz1, Sherry L. Jenkins1, John E. Evangelista1, Matteo Danieletto3, Jinshui Fan2, Mark W. Eshoo4‡, Michael R. Mosel4‡, William Robinson5, Nitya Ramadoss5, Jason Bobe3, Mark J. Soloski2, John N. Aucott2 and Avi Ma’ayan1
Department of Pharmacological Sciences, Mount Sinai Center for Bioinformatics, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Lyme Disease Research Center, Division of Rheumatology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
3Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Ibis Biosciences (an Abbott Laboratories company), Carlsbad, CA, United States
Division of Immunology and Rheumatology, Department of Medicine, Stanford University School of Medicine, Stanford, CA, United States
† These authors contributed equally to this work.
‡Present address: Mark W. Eshoo, BlueArc Biosciences, San Diego, CA, US. Michael R. Mosel, Janus-I Science Inc. Vista, CA, US
Publication Information:
Clarke DJB, Rebman AW, Bailey A, Wojciechowicz ML, Jenkins SL, Evangelista JE, Danieletto M, Fan J, Eshoo MW, Mosel MR, Robinson W, Ramadoss N, Bobe J, Soloski MJ, Aucott JN and Ma’ayan A (2021) Predicting Lyme Disease From Patients’ Peripheral Blood Mononuclear Cells Profiled With RNA-Sequencing. Front. Immunol. 12:636289. doi: 10.3389/fimmu.2021.636289
